References: 1. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141-1166. 2. Coman DJ, Hayes IM, Collins V, Sahhar M, Wraith JE, Delatycki MB. Enzyme replacement therapy and extended newborn screening for mucopolysaccharidoses: opinions of treating physicians. JIMD Rep. 2011;1:9-15. doi:10.1007/8904_2011_9. 3. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 4. Jurecka A, Zakharova E, Malinova V, Voskoboeva E, Tylki-Szymańska A. Attenuated osteoarticular phenotype of type VI mucopolysaccharidosis: a report of four patients and a review of the literature. Clin Rheumatol. 2014;33(5):725-731. doi:10.1007/s10067-013-2423-z. 5. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95-101. doi:10.1097/GIM.0b013e3181fea459. 6. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13-18. doi:10.1093/rheumatology/ker395. 7. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 8. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 9. Wood TC, Harvey K, Beck M, et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36(2):293-307. doi:10.1007/s10545-013-9587-1. 10. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 11. Thümler A, Miebach E, Lampe C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071-1079. doi:10.1007/s10545-012-9474-1. 12. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 13. Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30(2):165-174. doi:10.1007/s10545-007-0529-7. 14. Lachman RS, Burton BK, Clarke LA, et al. Mucopolysaccharidosis IVA (Morquio A syndrome) and VI (Maroteaux-Lamy syndrome): under-recognized and challenging to diagnose. Skeletal Radiol. 2014;43(3):359-369. doi:10.1007/s00256-013-1797-y. 15. Kinirons MJ, Nelson J. Dental findings in mucopolysaccharidosis type IV A (Morquio’s disease type A). Oral Surg Oral Med Oral Pathol. 1990;70(2):176-179. 16. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 17. Lachman R, Martin KW, Castro S, Basto MA, Adams A, Teles EL. Radiologic and neuroradiologic findings in the mucopolysaccharidoses. J Pediatr Rehabil Med. 2010;3(2):109-118. doi:10.3233/PRM-2010-0115. 18. Cimaz R, Coppa GV, Koné-Paut I, et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis [hypothesis]. Pediatr Rheumatol Online J. 2009;7:18. doi:10.1186/1546-0096-7-18. 19. Fahnehjelm KT, Ashworth JL, Pitz S, et al. Clinical guidelines for diagnosing and managing ocular manifestations in children with mucopolysaccharidosis. Acta Ophthalmol. 2012;90(7):595-602. doi:10.1111/j.1755-3768.2011.02280.x. 20. Zafeiriou DI, Batzios SP. Brain and spinal MR imaging findings in mucopolysaccharidoses: a review. AJNR Am J Neuroradiol. 2013;34(1):5-13. doi:10.3174/ajnr.A2832. 21. Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183-1197. doi:10.1007/s10545-011-9359-8. 22. Braunlin E, Orchard PJ, Whitley CB, Schroeder L, Reed RC, Manivel JC. Unexpected coronary artery findings in mucopolysaccharidosis. Report of four cases and literature review. Cardiovasc Pathol. 2014;23(3):145-151. doi:10.1016/j.carpath.2014.01.001. 23. Mesolella M, Cimmino M, Cantone E, et al. Management of otolaryngological manifestations in mucopolysaccharidoses: our experience. Acta Otorhinolaryngol Ital. 2013;33(4):267-272. 24. Berger KI, Fagondes SC, Giugliani R, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):201-210. doi:10.1007/s10545-012-9555-1. 25. Martins AM, Dualibi AP, Norato D, et al. Guidelines for the management of mucopolysaccharidosis type I. J Pediatr. 2009;155(4)(suppl 2):S32-S46. doi:10.1016/j.jpeds.2009.07.005. 26. Clarke LA, Winchester B, Giugliani R, Tylki-Szymańska A, Amartino H. Biomarkers for the mucopolysaccharidoses: discovery and clinical utility. Mol Genet Metab. 2012;106(4):396-402. doi:10.1016/j.ymgme.2012.05.003. 27. Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54-61. doi:10.1016/j.ymgme.2013.01.021. 28. Dhawale AA, Church C, Henley J, et al. Gait pattern and lower extremity alignment in children with Morquio syndrome. J Pediatr Orthop B. 2013;22(1):59-62. doi:10.1097/BPB.0b013e32835a0e6d. 29. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63-72. doi:10.1016/j.ymgme.2013.11.015. 30. Bhattacharya K, Balasubramaniam S, Choy YS, et al. Overcoming the barriers to diagnosis of Morquio A syndrome. Orphanet J Rare Dis. 2014;9:192. doi:10.1186/s13023-014-0192-7. 31. Choy YS, Bhattacharya K, Balasubramaniam S, et al. Identifying the need for a multidisciplinary approach for early recognition of mucopolysaccharidosis VI (MPS VI). Mol Genet Metab. 2015;115(1):41-47. doi:10.1016/j.ymgme.2015.03.005. 32. Data on file. Biomarin Pharmaceutical, Inc. 33. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 34. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78.



















{kind=link}
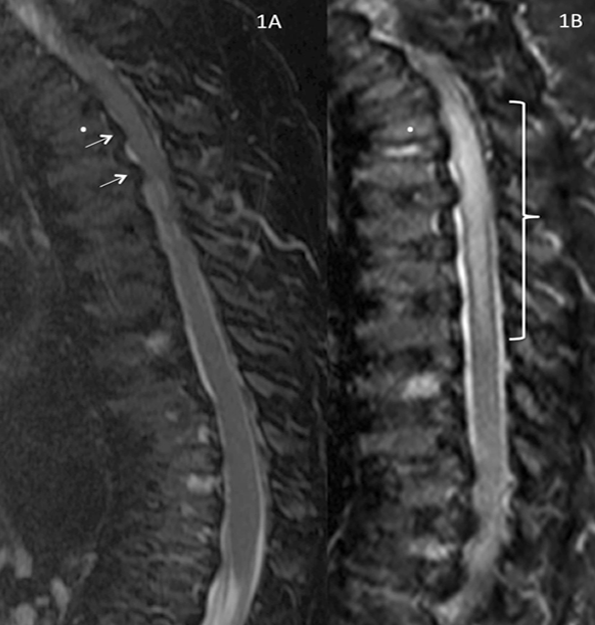{kind=link}
{kind=link}
{kind=link}