Décadas de investigación continua y experiencia clínica generaron una nueva era en la atención óptima de mucopolisacaridosis (MPS). Este estándar de cuidado de evolución rápida para MPS está basado en el genetista del centro del hogar médico—un modelo de atención de salud que comprende una atención coordinada, multidisciplinaria y le brinda a los médicos una oportunidad inigualable de cambiar la vida de los pacientes.1-3
Esta naturaleza heterogénea y variable de las enfermedades MPS requiere un enfoque personalizado para una atención coordinada del paciente, que comienza en el hogar médico.4 El objetivo de la atención compaginada es ayudar a los pacientes a alcanzar una mejor calidad de vida, que incluye:
Para los pacientes pediátricos con enfermedad genética crónica, compleja y multisistémica como MPS, la atención a través de un hogar médico coordinado está asociada con un menor uso de atención médica y mejores resultados.5-8
La coordinación dentro del hogar médico debe implementarse en todos los elementos del amplio sistema de salud (ej. cuidado especializado, hospitales, atención domiciliaria y servicios de la comunidad) y dentro de los planes de atención personalizados de cada paciente.3
La aplicación del cuidado óptimo para MPS, agrupado en los siguientes pilares de atención, permiten mejorar los resultados de los pacientes :

En paralelo al hogar médico, los planes de atención personalizados que incluyen TRE, atención de por vida y cuidado del paciente quirúrgico ayudan a optimizar los resultados de los pacientes.2,11,13
La TRE, cuando se encuentra disponible, es el pilar fundamental de la terapia.12-14
Un paciente con MPS VI de progresión rápida es detectado antes de tiempo, posibilitando el inicio inmediato de TRE y la obtención de mejores resultados clínicos.20

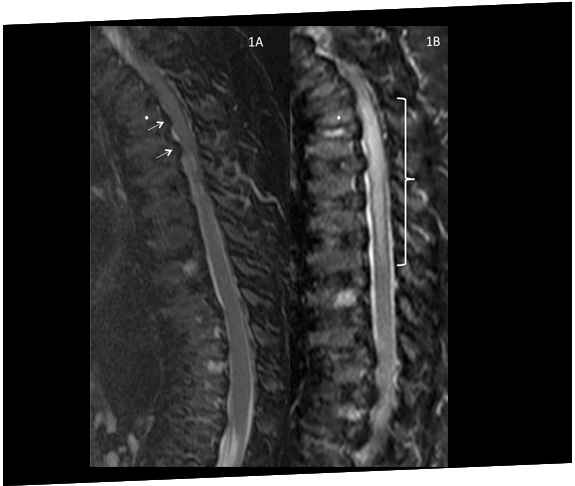
Un paciente con Morquio A (MPS IVA) desarrolla compresión espinal y paraplejía inmediatamente después de una cirugía para corregir deformidades genu valgo, destacando la vulnerabilidad a la compresión durante la anestesia.21
Un niño con retrasos en el desarrollo con síntomas percibidos durante un período de 7 años recibe posteriormente un diagnóstico de MPS VI y comenzó con TRE, mostrando mejoras clínicamente significativas tras la TRE.20
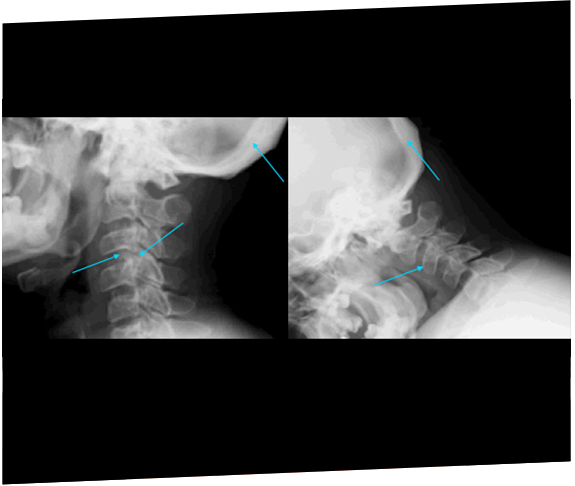
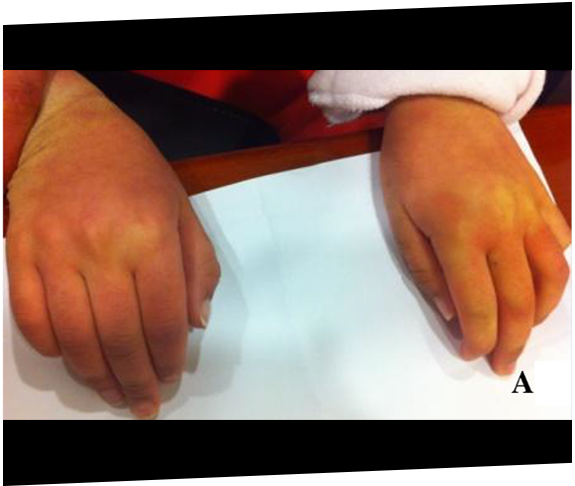
Dos hermanas fueron finalmente diagnosticadas con MPS IIIA después de los 11 años como consecuencia de una progresión rápida del retraso de desarrollo y la posterior derivación a un genetista.22
A pesar de que cada subtipo de MPS es clínicamente diferente, todos poseen las manifestaciones limitantes para la vida, progresivas, multisistémicas comunes de la patología de enfermedades MPS.14,16,23,24 La atención de pacientes con MPS requiere un conocimiento específico de las manifestaciones clínicas y de las recomendaciones de cuidado para cada subtipo de MPS.2,13
noviembre de, 2015
No se alcanzó analgesia bilateral óptima en el trabajo de parto a pesar de múltiples ajustes y se necesitó analgesia sistémica para cesárea.
mayo de, 2015
Demoras en el diagnóstico ocurrieron debido a la falta o al alejamiento de unidades diagnósticas, diagnósticos alternativos y presentación de síntomas que indujeron a errores. Muchos pacientes experimentaron manifestaciones más sutiles que las que se esperarían y, las que posteriormente pasaron inadvertidas. Los casos también destacaron los desafíos únicos asociados con el diagnóstico de MPS VI desde el punto de vista de diferentes especialidades y brindan información sobre como estos pacientes se presentan al principio.
abril de, 2016
Nuestro conocimiento actual de otros problemas cardíacos en adultos con las MPSs, en especial con la circulación coronaria al miocardio, es escaso y hay que estudiarlos más para brindar atención eficaz a esta población creciente de adultos.
References: 1. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 3. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 4. Hendriksz CJ, Harmatz P, Beck M, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54-64. doi:10.1016/j.ymgme.2013.04.002. 5. Casey PH, Lyle RE, Bird TM, et al. Effect of hospital-based comprehensive care clinic on health costs for Medicaid-insured medically complex children. Arch Pediatr Adolesc Med. 2011;165(5):392-398. doi:10.1001/archpediatrics.2011.5. 6. Mosquera RA, Avritscher EBC, Samuels CL, et al. Effect of an enhanced medical home on serious illness and cost of care among high-risk children with chronic illness: a randomized clinical trial. JAMA. 2014;312(24):2640-2648. doi:10.1001/jama.2014.16419. 7. Klitzner TS, Rabbitt LA, Chang RKR. Benefits of care coordination for children with complex disease: a pilot medical home project in a resident teaching clinic. J Pediatr. 2010;156(6):1006-1010. doi:10.1016/j.jpeds.2009.12.012. 8. Gordon JB, Colby HH, Bartelt T, Jablonski D, Krauthoefer ML, Havens P. A tertiary care-primary care partnership model for medically complex and fragile children and youth with special health care needs. Arch Pediatr Adolesc Med. 2007;161(10):937-944. 9. Chiang J, Raiman J, Cutz E, Solomon M, Dell S. Tachypnea of infancy as the first sign of Sanfilippo syndrome. Pediatrics. 2014;134(3):e884-e888. doi:10.1542/peds.2013-2765. 10. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12(2):133-138. doi:10.1016/j.prrv.2010.10.005. 11. Berger KI, Fagondes SC, Giugliani R, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):201-210. doi:10.1007/s10545-012-9555-1. 12. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 13. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. 14. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95-101. doi:10.1097/GIM.0b013e3181fea459. 15. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141-1166. 16. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 17. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. doi:10.1007/8904_2014_298. 18. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584. 19. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983. 20. Data on file. Biomarin Pharmaceutical, Inc. 21. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 22. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78. 23. Clarke LA, Winchester B, Giugliani R, Tylki-Szymańska A, Amartino H. Biomarkers for the mucopolysaccharidoses: discovery and clinical utility. Mol Genet Metab. 2012;106(4):396-402. doi:10.1016/j.ymgme.2012.05.003. 24. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 25. Ashworth JL, Biswas S, Wraith E, Lloyd IC. Mucopolysaccharidoses and the eye. Surv Ophthalmol. 2006;51(1):1-17.